
CXC Home | Search | Help | Image Use Policy | Latest Images | Privacy | Accessibility | Glossary | Q&A
The Astronomers Tycho Brahe and Johannes Kepler

Tycho Brahe
Tycho Brahe (1546-1601) was a nobleman from Denmark who made astronomy his life's work because he was so impressed when, as a boy, he saw an eclipse of the Sun take place at exactly the time it was predicted. Tycho's life's work in astronomy consisted of measuring the positions of the stars, planets, Moon, and Sun, every night and day possible, and carefully recording these measurements, year after year.

Johannes Kepler
Johannes Kepler (1571-1630) came from a poor German family. He did not have it easy growing up. His father was a soldier, who was killed in a war, and his mother (who was once accused of witchcraft) did not treat him well. Kepler was taken out of school when he was a boy so that he could make money for the family by working as a waiter in an inn. As a young man Kepler studied theology and science, and discovered that he liked science better. He became an accomplished mathematician and a persistent and determined calculator. He was driven to find an explanation for order in the universe. He was convinced that the order of the planets and their movement through the sky could be explained through mathematical calculation and careful thinking.
Kepler's De Stella Nova, 1606
Tycho wanted to study science so that he could learn how to predict eclipses. He studied mathematics and astronomy in Germany. Then, in 1571, when he was 25, Tycho built his own observatory on an island (the King of Denmark gave him the island and some additional money just for that purpose). Tycho named his island observatory Uraniburg-Urania after the muse of astronomy. He lived and worked in his observatory until he had a disagreement with the King of Denmark. Tycho’s main goal was to determine the positions of the stars and planets as accurately as possible. This could only be done by constructing precision observing instruments and making and recording many observations of stars and planets night after night.
Kepler became interested in science and mathematics when in school at about the age of 18. He was not particularly interested in astronomy until 1600 when Kepler met Tycho Brahe in Prague, and Tycho asked him to be his assistant. Tycho would pay him well. However, Tycho died one year later, and even though Kepler was appointed astronomer to the court, he found so little official support for his position that he had to survive by making astrological predictions for noblemen who wanted their fortunes told.

Uraniborg – Tycho's Observatory in Denmark
Tycho was a scientist who worked by direct observation. Kepler was a scientist who worked by calculation and testing one idea after another. Tycho's life's work of measuring the positions of objects in the sky was in itself useless without someone like Kepler to come along and make sense of those measurements. In the same way, Kepler's efforts to understand how the planets moved would be nothing but speculation, guessing, and mysticism if he did not have the basic data – the accurate measurements made by Tycho – against which to test his ideas and theories. Each one’s work is meaningful because of the work of the other. The contributions to science by these two astronomers from radically different backgrounds was set against a time of great turmoil in European history – the early 1600's. It was a time of upheaval, superstition, and fear – a time when court astrologers were powerful, and the stars were thought to predict and guide one's destiny.

Tycho Brahe’s Skeleton, November, 2010
After Brahe's difficulties with the King of Denmark, he obtained the position of Royal Mathematician at the court-in-exile of the Holy Roman Emperor, Rudolf II, in Prague. He was arrogant, conceited, and obnoxious. While at university he had a duel with a fellow student over which one was the best mathematician. Tycho may have been the superior mathematician, but he was not the better duelist: during the encounter he lost his nose, which he replaced with one made of gold. He had different metal noses which he changed depending upon the occasion. At a dinner given by a local Baron, Tycho consumed great quantities of wine but would not leave the table in the presence of the Baron, considering it to be rude behavior. It has long been thought that the resulting urinary tract infection, along with Tycho's refusal to stop abusing his body with overdrinking and overeating, might have led to his death a few days later. According to medical experts however, bladder ruptures are rare, and Brahe probably died from kidney failure. The first exhumation of Tycho's remains at Tyne Church in Prague was for the purpose of determining his cause of death. The exhumation produced new questions about Tycho Brahe’s death – including the possibility of murder!

Tycho Brahe’s Skull, November, 2010
On November 10, 2010, the remains of Tycho Brahe were once again exhumed. The remains were transported to Prague to be examined by an international team of Danish and Czech archaeologists, doctors, chemists and medical anthropologists. The researchers hope to use DNA testing and other modern medical diagnostic tools to learn as much as possible about Brahe's medical history, as well as his cause of death. In 1996 tests were conducted on samples of hair that had been taken from Brahe’s body the first time it was exhumed, back in 1901. Those tests indicated high levels of mercury, and that in the 24 hours before Brahe died he had ingested a large amount of mercury – though the test did not indicate either the actual amount of mercury or if the amount was fatal. Tycho Brahe died in 1601 at the age of 54.

Kepler's Astronomica Nova, 1906
Did someone administer the mercury to Brahe? Did Johannes Kepler kill Brahe to get his hands on Brahe’s observations? Owen Gingerich, who is a professor of astronomy at Harvard, does not agree with this theory. He is an expert on Kepler, and he stated that it would not make sense for Kepler to kill Tycho because at the time he died, Tycho was trying to convince the emperor to make Kepler the imperial mathematician. By killing Tycho, Kepler would have ruined that opportunity. However, Kepler did get the job even after Tycho’s death. Did the Danish King Christain IV order Brahe’s poisoning because Brahe had slept with the king’s mother? Did Tycho – who was also an alchemist, accidently ingest mercury during one of his experiments? Or did he suffer a fatal overdose of mercury while self-medicating for his painful kidney ailment? The test results from the 2010 exhumation are expected to contribute to knowledge of his life and perhaps his cause of death – so far those results have not been released to the public as of December, 2011).

Kepler and Rudolph II in Prague
Johannes Kepler was born into much humbler surroundings. He expected to enter the clergy, but instead became a mathematics teacher in Graz, Austria. His belief in the Copernican concept of a heliocentric universe was a dangerous one. With the coming of the 30 Years' War, Kepler and his wife were exiled due to their Protestant beliefs. During Kepler’s time in Prague working as Tycho’s assistant, they fought continuously, because Tycho refused to share his meticulous observations with Kepler. These were observations which Kepler desperately needed for his continuing quest to establish the true orbital motions of the planets. After Tycho’s death, Kepler stole the data in order to continue his calculations.

The Somnium

Bettina Forget Artwork Based on The Somnium
Eventually the war reached Prague, and Kepler was once again persecuted for his religious beliefs. He also lost his wife and son to a plague, and his mother was convicted of witchcraft and imprisoned. It took Kepler five years to get his mother released and her sentence commuted to one of exile. During this time Kepler wrote what many consider to be the first work of science fiction, entitled Somnium (The Dream). This story probably contributed to his mother's persecution as she resembled one of the characters – an old woman who had dealings with demons and devils. Kepler tells the tale through the eyes of a young man who bears obvious similarities to Kepler himself. So when the story hints that the protagonist’s mother is a witch; authorities make a connection with Kepler’s real-life mother and arrest her on charges of being a witch. It didn’t help that, years before, the woman who raised Kepler’s mother had actually been burned as a witch. Somnium is an incredible story for an astronomer and mathematician in the early 1600’s – full of moon people, space travel, and magical beings. What an imagination! The lunar inhabitants weren’t mere recreations of terrestrial life, but entirely new forms of life adapted to lunar extremes. Large and tough-skinned, they evoke visions of dinosaurs. Some used boats, implying not just life but intelligent, non-human life. Imagine how shocking that must have been at the time. Kepler still was not done with the Somnium. After his mother died, Kepler completed a set of explanatory footnotes which by themselves were almost three times longer than the text. He intended to publish the Somnium bound with ancient works by Plutarch and Lucian — books that both inspired Kepler’s efforts and would have lent credibility to the Somnium. Unfortunately, he died before he could realize his "Dream." The final version of the Somnium was published shortly after his death as a stand-alone work. Exactly what Kepler intended when he wrote the Somnium is not clear, and the work remains a bit of a puzzle. But the result, the mix of facts and enlightened dreaming, are hallmarks of classic science fiction.
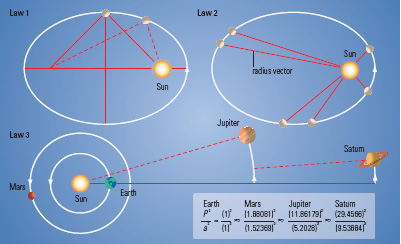
Kepler's 3 Laws of Planetary Motion
Kepler finished his days in poverty, writing horoscopes for noblemen in order to survive. Tycho Brahe and Johannes Kepler had totally disparate backgrounds and temperaments. In spite of this, Tycho's painstaking and detailed observational data of the planet Mars, combined with Kepler's mathematical genius, allowed Kepler to derive the three laws of planetary motion. Both Tycho and Kepler made significant contributions to the change in the prevailing world view of a geocentric universe. It was the beginning of a systematic study that transformed Medieval thinking – alchemy became chemistry and astrology led to astronomy.
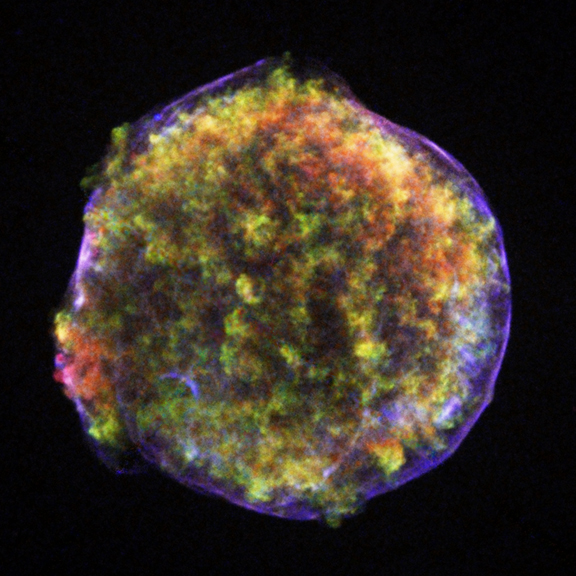
The Tycho and Kepler Supernova ObservationsTycho Brahe was walking home from his laboratory on November 11, 1572 when his attention was attracted by the star in the constellation of Cassiopeia which was as bright as Jupiter and had not been visible before. Tycho wrote the following description (from Burnham’s Celestial Handbook):
"On the 11th day of November in the evening after sunset, I was contemplating the stars in a clear sky. I noticed that a new and unusual star, surpassing the other stars in brilliancy, was shining almost directly above my head; and since I had, from boyhood, known all the stars of the heavens perfectly, it was quite evident to me that there had never been any star in that place of the sky, even the smallest, to say nothing of a star so conspicuous and bright as this. I was so astonished of this sight that I was not ashamed to doubt the trustworthiness of my own eyes. When I observed that others, on having the place pointed out to them, could see that there was really a star there, I had no further doubts. A miracle indeed, one that has never been previously seen before our time, in any age since the beginning of the world."

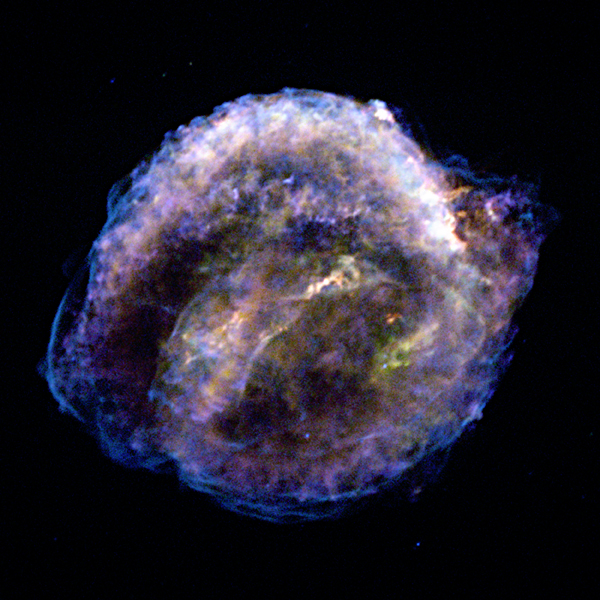
De Stella Nova, Prague, 1606
Most people assume that because the supernova of 1604 was named after Kepler, that he must have been the first to see it. However, historical reports indicate that the supernova was first seen in northern Italy on the evening of October 9, 1604, and by the Chinese and Koreans during the next few days. In Prague, an independent sighting was made on October 10th through a break in the clouds, by J. Brunowski, who reported the sighting to Kepler. Cloudy weather in Prague prevented Kepler from observing the object until the evening of October 17th. Kepler observed the newly visible star over the course of a year, and in 1606 published a detailed account in his book – De Stella Nova.
These two supernova events were observed and recorded; the dates can not be disputed. Both were Type Ia supernova events – the thermonuclear explosion of a white dwarf approaching the Chandrasekhar limit. Both were close enough to Earth that they were observable with the naked eye. Both supernovas were observed and studied until they faded from view several months later. It seems fitting – considering the interconnected lives of Tycho and Kepler –they would both have a supernova named after them.

Statue of Johannes Kepler (left) and Tycho Brahe (right) in Prague
Back to Ice Core Index
Revised: January 30, 2012